
Total de visitas: 106349
|
SÍNDROME DE KLINEFELTER
Síndrome de Klinefelter ( 47,XXY)
O Dr. Harry F. Klinefelter ao trabalhar no projeto de consumo de oxigênio na glândula adrenal em conjunto com o Dr. Howard Means atendeu um paciente com um caso raro no qual um homem desenvolveu seios (Ginecomastia).
Ao estudar este caso, o Dr. Klinefelter relatou nos seu exames infertilidade, liberação de hormônio Gonadotropina (GnRH), um elevado nível de liberação de Hormônio Folículo Estimulante (FSH) e Hormônio Luteneizante (LH).
Com estes resultados foi publicado no Jornal de Metabolismo e Endocrinologia Clínica (1942), um artigo intitulado Síndrome caracterizada por Ginecomastia, aspermatogênese e aumento da excreção de Hormônio Folículo Estimulante, tendo como autores, Klinefelter H G, Reifestein E C Jr., e Albright F..
Desde então, a literatura só chama esta condição de síndrome de Klinefelter (SK).
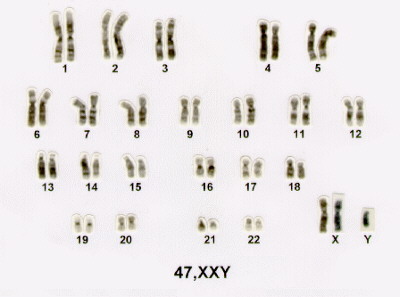
A SK é causada por uma variação cromossômica envolvendo o cromossomo sexual. Este cromossomo sexual extra (X) causa uma mudança característica nos meninos. Todos os homens possuem um cromossomo X e um Y, mas ocasionalmente uma variação irá resultar em um homem com um X a mais, esta síndrome é muitas vezes escrita como 47 XXY. Existem outras variações menos comuns como: 48 XXYY; 48 XXXY; 49 XXXXY; e mosaico 46 XY/47 XXY, este é o cariótipo mais comum, ocorre em cerca de 15% provavelmente em conseqüência da perda de um cromossomo X num concepto XXY durante uma divisão pós-zigótica inicial.
Metade dos casos resulta de erros na meiose I paterna, um terço de erros na meiose I materna e os demais de erros na meiose II ou de um erro mitótico pós-zigótico levando a mosaico. A idade da mãe é elevada nos casos associados a erros na meiose I materna, mas não nos outros casos (JACOBS et al., 1988).
Até 1960 o diagnóstico era feito através de exame histológico dos testículos que, mesmo após a puberdade, revelava ausência de células germinativas nos canais seminíferos. Atualmente a identificação dos Klinefelter é assegurada pelo cariótipo e pela pesquisa da cromatina sexual, através de um exame feito com uma amostra de sangue.
As estatísticas mostram que a cada 500 nascimentos é encontrado um menino com a síndrome.
Cariótipo
Características do Portador
A característica mais comum é a esterilidade. Possuem função sexual normal, mas não podem produzir espermatozóides (Azoospermia) devido à atrofia dos canais seminíferos e, portanto são inférteis.
Outras características muitas vezes presentes são: estatura elevada e magros, com braços relativamente longos; pênis pequeno; testículos pouco desenvolvidos devido à esclerose e hialinização dos túbulos seminíferos ; pouca pilosidade no púbis; níveis elevados de LH e FSH, podem apresentar uma diminuição no crescimento de barba; ginecomastia (crescimento das mamas), devido aos níveis de estrogênio (hormônio feminino) mais elevados do que os de testosterona (hormônio masculino). Em alguns casos tornam-se necessária à remoção cirúrgica; problemas no desenvolvimento da personalidade provavelmente em decorrência de uma dificuldade para falar que contribuem para problemas sociais e/ou aprendizagem.
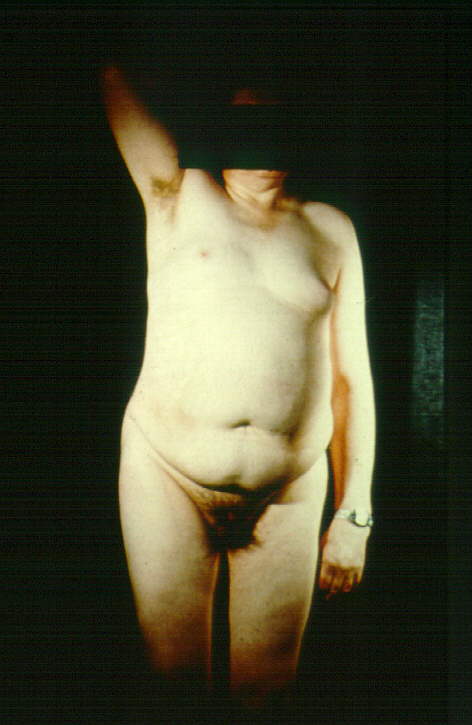

Ginecomastia

Alta Estatura
Tratamento
Deve ser feito o acompanhamento periodico do nível de testosterona (hormônio sexual masculino) no sangue, para verificar sua normalidade. Caso o nível de testosterona encontre-se baixo isso irá resultar na diminuição das mudanças sexuais que ocorrem durante a puberdade.
Para controle é comum a aplicação de uma vez ao mês uma injeção de Depotestosterona, uma forma sintética de testosterona. A dose necessita ser aumentada gradualmente e ser aplicada mais freqüentemente quando o menino torna-se mais velho.
XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
MAIS TEXTOS SOBRE ESSA SÍNDROME:
Síndrome de Klinefelter
O termo síndrome de Klinefelter (SK) descreve um grupo de anomalias cromossómicas, nas quais há, pelo menos, um cromossoma X adicional ao cariótipo masculino normal, 46,XY. A aneuploidia XXY é a anomalia mais comum dos cromossomas sexuais, com uma frequência de 1:500 indivíduos. As variantes do síndrome de Klinefelter são muito menos frequentes, tendo os 48,XXYY e os 48,XXXY uma incidência de 1 por 50,000 nados-vivos do sexo masculino. A incidência de 49,XXXXY é 1:85.000-100.000 nados-vivos do sexo masculino e é a variante mais grave deste síndrome. O cariótipo é essencial para fazer um diagnóstico definitivo. Um número crescente de cromossomas sexuais tende a estar associado a anomalias físicas mais marcadas. A análise cromossómica em linfócitos de sangue periférico, amniócitos ou vilosidades coriónicas (diagnóstico pré-natal) é utilizada para fazer este diagnóstico. Se o diagnóstico não for feito no período pré-natal, os indivíduos 47,XXY podem apresentar uma diversos sinais clínicos subtis que estão relacionados com a idade. Durante os primeiros anos de vida, o diagnóstico de S. Klinfelter pode ser feito através do cariótipo que foi pedido na investigação de hipospádias, micropénis ou criptorquidia. As crianças em idade escolar podem apresentar atraso da linguagem, problemas de aprendizagem ou do comportamento. As crianças mais velhas ou adolescentes podem ser diagnosticadas durante avaliação endocrinológica por atraso do desenvolvimento pubertário com habito eunucóide, ginecomastia e testículos pequenos. Os adultos são muitas vezes avaliados por infertilidade ou neoplasia mamária. A terapia de substituição com androgénios deve ser iniciada na puberdade, cerca dos 12 anos, numa dose crescente, que deverá ser suficiente para manter concentrações séricas apropriadas de testosterona, estradiol, FSH e LH. A terapia com androgénios promove a normalização das proporções corporais e o normal desenvolvimento dos caracteres sexuais secundários, ajudando a prevenir a ginecomastia. No síndrome de Klinfelter, o cromossoma X extra surge esporadicamente como resultado de não-disjunção meiótica (quando um cromossoma não se separa durante a primeira ou segunda divisão da gametogénese) ou por não-disjunção mitótica (durante o desenvolvimento do zigoto). A probabilidade de não-disjunção do cromossoma X aumenta com a idade materna. Os efeitos no desenvolvimento mental e físico aumentam com o número de cromossomas X extra, sendo que, por cada um destes se verifica uma diminuição no QI de 15-16 pontos. A linguagem é principalmente afectada, sobretudo as capacidades expressivas. *Autores: Dr. J Vissotsak e Prof. JM Graham (Março 2003).*
Alta estatura
Anomalia cromossómica numérica
Criptorquidia/testículos ectópicos
Esterilidade/hipofertilidade
Membros longos
Micropénis/pénis pequeno
Puberdade tardia/hipogonadismo
Anomalias dos dermatoglifos excluindo prega palmar
Clinodactilia do quinto dedo
Doença do movimento
Escoliose
Ginecomastia/hipertrofia mamária
Obesidade generalizada
Osteoporose
Prega palmar
Alteração do comportamento/autismo
Atraso mental (ligeiro)
Braquicefalia/achatamento occipital
Hiperglicemia/diabetes mellitus
Hipospádias/epispádias
MAIS IMAGENS:
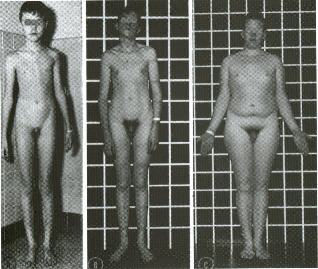
Indivíduos XXY

XXXY XXXY XXXXY



|
|

