
Total de visitas: 106665
|
SÍNDROME DE PATAU
Síndrome de Patau ou Trissomia do 13
Reconhecida em 1960 por Klaus Patau observando um caso de malformações múltiplas em um neonato, sendo trissômico para o cromossomo 13. Tem como causa a não disjunção dos cromossomos durante a anáfase 1 da mitose, gerando gametas com 24 cromátides. Cerca de 20% dos casos resultam de uma translocação não-balanceada.
A sua incidência foi estimada em cerca de 1 caso para 6000 nascimentos. Aproximadamente 45% dos afetados falecem após 1 mês de vida; 70%, aos 6 meses e somente menos de 5% dos casos sobrevivem mais de 3 anos. A maior sobrevida relatada na literatura foi a de 10 anos de idade.
Assim como a maioria das outras trissomias, associa-se à idade materna avançada, por estarem mais propícias a ocorrência da não disjunção dos cromossomos. A idade da mãe é superior a 35 anos em 40% dos casos.
A trissomia tem origem do óvulo feminino, pelo fato da fêmea maturar geralmente apenas um ovócito, em antagonismo com o macho, que matura milhões de espermatozóides. Gametas masculinos portadores de alterações numéricas cromossômicas tem menor viabilidade que gametas normais, sendo mínimas as possibilidades de um gameta masculino com 24 cromátides fecundar um ovócito.
Cariótipo
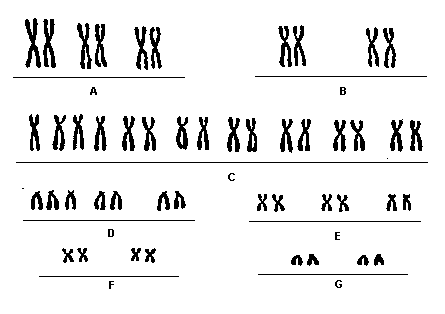
Característica dos Portadores
O fenótipo inclui malformações graves do sistema nervoso central como arrinencefalia. Um retardamento mental acentuado está presente. Em geral há defeitos cardíacos congênitos e defeitos urigenitais incluindo criptorquidia nos meninos, útero bicornado e ovários hipoplásticos nas meninas gerando inviabilidade, e rins policísticos. Com freqüência encontram-se fendas labial e palato fendido, os punhos cerrados e as plantas arqueadas. A fronte é oblíqua, há hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia, coloboma da íris, olhos são pequenos extremamente afastados ou ausentes. As orelhas são malformadas e baixamente implantadas. As mãos e pés podem mostrar quinto dedo (polidactilia) sobrepondo-se ao terceiro e quarto, como na trissomia do 18.
Fenda Labial

XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
OUTRA MATÉRIA SOBRE SÍNDROME DE PATAU
Trissomia do cromossomo 13 ou síndrome de Patau
1 . Trissomia do 13:
A trissomia do cromossomo 13 tem como definição uma desordem cromossomal resultando em uma síndrome caracterizada especificamente pôr anomalias morfológicas e malformação de órgãos, gerando a inviabilidade dos afetados. Ocorre quando existem três cromossomos 13 em lugar do par normal no genótipo de um recém nascido. Tem como causa a não disjunção dos cromossomos durante a anáfase 1, gerando gametas com 24 cromátides.
2. Histórico:
Observada na literatura a primeira vez em 1657 pôr Bartholin, e descrita em 1960 pôr Patau e colaboradores, que a denominaram trissomia do cromossomo D1. Logo em seguida, a síndrome determinada pôr essa aneuploidia foi minuciosamente estudada pôr diversos autores, de sorte que, e, em pouco tempo, ela pôde ser caracterizada clinicamente com bastante precisão. Estudos auto-radiográficos e de fluorescência forneceram evidências de que o cromossomo trissômico nesta síndrome é o 13.
3. Epidemiologia:
Ocorre na variação de 1/4.00010.000 crianças que nascem, sendo letal geralmente no primeiro mês da doença. Tem a probabilidade de risco agravada pôr uma possível gravidez tardia (>37 anos), devido ao fato de que mulheres acima desta idade, estarem mais propícias a ocorrência da não disjunção dos cromossomos. Parece possuir ligeira preferência pelo sexo feminino.
4. Patogênese:
Genética:
Trissomia do 13: Quadro clínico rico em sinais e cerca de 75% dos casos apresentam cariótipo com trissomia regular. A trissomia é devida ao fato de não haver disjunção dos cromossomos durante a anáfase 1 da mitose. A gravidez em idade avançada aumenta a possibilidade da não disjunção dos cromossomos. Sua taxa de ocorrência é 1/4.000.
Nas figuras 1 e 2 é possível ver o cariótipo de um afetado e um esquema mostrando a primeira não disjunção meiótica numa mulher respectivamente:
Fig 1.: Cariograma de um afetado pela trissomia do 13
A trissomia tem origem do óvulo feminino, pelo fato da fêmea maturar geralmente apenas um ovócito, em antagonismo com o macho, que matura milhões de espermatozóides. Gametas masculinos portadores de alterações numéricas cromossômicas tem menor viabilidade que gametas normais, sendo mínimas as possibilidades de um gameta masculino com 24 cromátides fecundar um ovócito.
Translocações: São responsável pôr 20% das ocorrências, sendo que ¾ das translocações são esporádicas, e ¼ são herdadas. As translocações familiares balanceadas ou não envolvendo cromossomos do grupo D [mais freqüentemente t(13q14q)] estão entre as mais comuns, dentre as variações da síndrome. As pessoas normais que tem essa translocação equilibrada estão sob o risco de 5% não apenas de gerar uma criança com síndrome de Patau, mas também de ter em sua prole crianças com síndrome de Down com trissomia regular, pôr um efeito intercromossômico.
Mosaicismo: Representa 5% das ocorrências da síndrome, sendo verificado pelo fato do afetado não possuir a anomalia cromossômica de forma detectável. São representados da seguinte maneira: 46,XX ou XY/47, XX ou XY, + 13. Esta alteração pode propiciar que o afetado chegue até a idade adulta, mesmo manifestando o fenótipo.
Patogênese: Um problema durante as 3 primeiras semanas de desenvolvimento do embrião, na diferenciação do mesoderma, na placa pré-cordal, gera a anomalia no indivíduo que possui o genótipo.
5. Diagnóstico:
O diagnóstico clínico da síndrome de Patau, a ser confirmado pelo exame cromossômico é fácil, pois dentre os seus sinais mais comuns estão:
- baixo peso corporal (2.600Kg);
- Microcefalia e testa achatada;
- Largas suturas sagitais e fontanelas;
- Hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia;
- Lábio leporino acompanhado ou não de palatosquise ou palato alto;
- Queixo pequeno;
- Defeitos na face média e cérebro anterior;
- Orelhas dismórficas de implantação baixa e surdez aparente;
- Pescoço curto;
- Fronte inclinada;
- Hemangiomas planos na cabeça;
- Distância intermamilar grande;
- Apnéias prolongadas;
- Cardiopatias congênitas, que representam comunicação interventricular e persistência do conduto arterial;
- Apêndice pré-sacral e fóvea coccigeana;
- Hérnia inguinal ou umbilical;
- Genitais externos anômalos (criptorquidia escroto, abdominal, genitália ambígua e pênis encurvado entre os meninos, e clitoromegalia e vagina dupla entre as meninas);
- Mãos com hexadactilia uni- ou bilateral, geralmente com o polegar e os dois últimos dedos sobrepostos aos outros; unhas estreitas e hiperconvexas;
- Prega de flexão palmar única, trirrádio axial em posição bastante distal (t e t ) e arco na região tenar;
- Pés com hexadactilia uni- ou bilateral e com região plantar convexa (pés em cadeira de balanço);
- Arco ou arco retorcido em S na região halucal.
- Arrinencefalia (ausência de bulbo e trato olfativo);
- Deficiência mental;
- Útero bicorne;
- Rim policístico, hidronefrose, hidroureter e ureteres duplos, relacionados a oligúria e anúria em afetados;
- Atrofia ou ausência das últimas costelas e de vértebras, e hiperplasia sacral;
- Presença de Hemoglobina Gower 2, que é uma hemoglobina embrionária que desaparece no terceiro mês de gestação;
- Neutrófilos com núcleo mostrando muitas saliências pedunculadas ou sésseis;
Crianças afetadas pela síndrome de Patau:
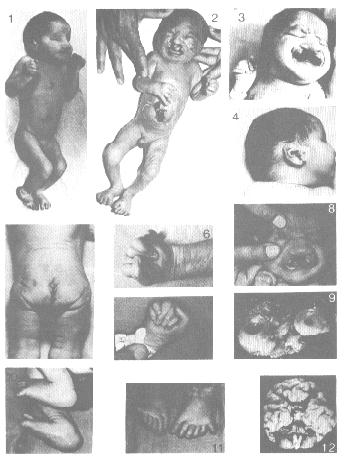
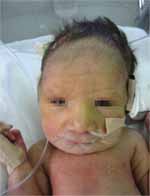

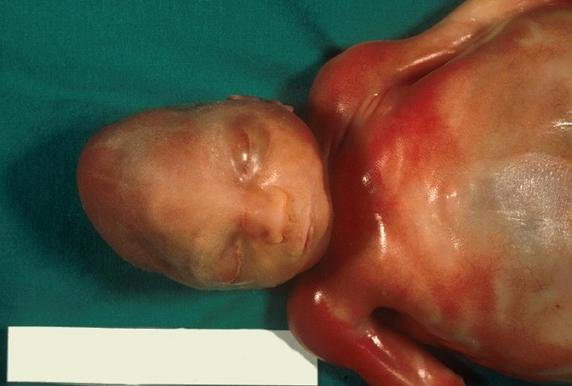

6. Investigações:
- Para observar más formações:
o Anomalias cardiovasculares: Echo;
o Anomalias no sistema nervoso central: CT/MRI;
o Anomalias genito-urinárias: Ultrasom;
- Cariótipo
- Amniocinese
7. Tempo de sobrevivência dos afetados:
45% morrem em 1 mês;
69% morrem em 6 meses;
72% morrem em 1 ano;
8. Aconselhamento médico:
Genitores devem ser informados de que a trissomia do 13 tem a taxa de ocorrência de 1/4.000 10.000;
Necessidade de avaliar o cariótipo dos genitores para saber a probabilidade de chances de ocorrer um afetado;
No caso de uma gravidez com ocorrência da síndrome, é necessário acompanhamento com amniocinese na gestações subsequentes.
|
|

